For individuals with rare diseases, getting a diagnosis is often a long and complicated odyssey. Over the past few years, this has been greatly improved by genome sequencing that can pinpoint the mutation that breaks a gene and leads to a severe disease. However, this approach is still unsuccessful in the majority of patients, largely because of our inability to read the genome to identify all mutations that disrupt gene function.
In a new study published on October 10 in Science, researchers from New York Genome Center, Columbia University, and Scripps Research Institute propose a solution to this problem. Building a new computational method for analyzing genomes together with transcriptome data from RNA-sequencing, they can now identify genes where genetic variants disrupt gene expression in patients and improve the diagnosis of rare genetic disease.
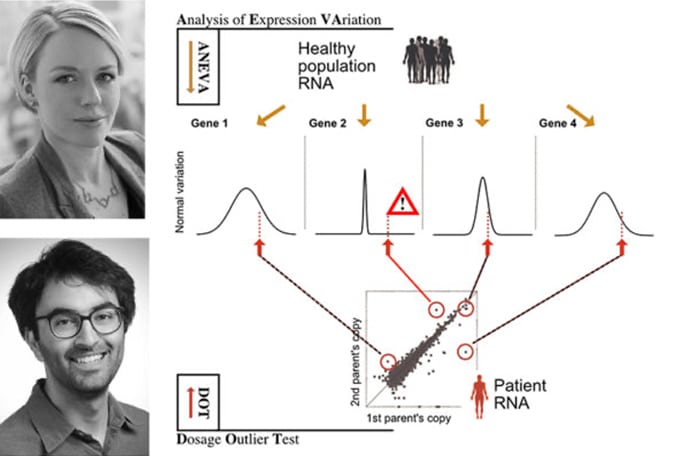
The new method introduced in this study, Analysis of Expression Variation or ANEVA, first takes allele-specific expression data from a large reference sample of healthy individuals to understand how much genetic regulatory variation each gene harbors in the normal population. Then, using the ANEVA Dosage Outlier Test, researchers can analyze the transcriptome of any individual – such as a patient – to find a handful of genes where he or she carries a genetic variant with an unusually large effect compared to what healthy individuals have. By applying this test to a cohort of muscle dystrophy and myopathy patients, the researchers demonstrated the performance of their method and diagnosed additional patients where previous methods of genome and RNA analysis had failed to find the broken genes.
The study was led by Pejman Mohammadi, PhD, a former postdoctoral scientist at the New York Genome Center and Columbia University and now an assistant professor at the Scripps Research Institute, and supervised by Tuuli Lappalainen, PhD, core faculty member at the New York Genome Center and an assistant professor at Columbia University. The illustration above depicts with an example of four genes, how knowing how variable genes are in the normal population helps to find candidate disease genes in a patient. Their method is implemented in software released with the paper and can be applied to any rare disease patients where RNA-sequencing and genetic data exists.


