A multi-institutional cancer genomics research team, led by Marcin Imielinski, MD, PhD, Core Faculty Member at the New York Genome Center (NYGC), has released its findings from a comprehensive whole-genome sequencing (WGS) study of a key subset of lung adenocarcinoma. The team’s report, published today in Cell Reports, uncovers structural genetic variants that may be potential new biomarkers and therapeutic targets for this subset as well as advances understanding of the mechanisms of LUAD disease. LUAD is the most common lung malignancy and a leading cause of cancer death in the U.S.
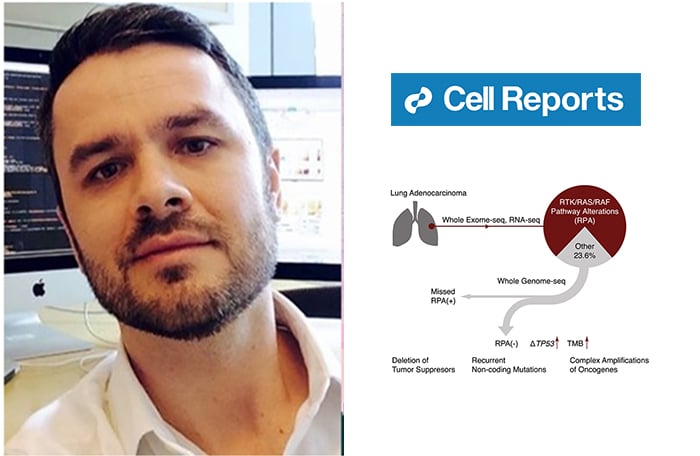
Specifically, the research team analyzed LUAD cancer tumor samples with no evidence of RTK/RAS/RAF pathway alterations, known as “RPA negative cancers”. Over the past decade, genomic studies, including several studies co-led by Dr. Imielinski, have identified that RTK/RAS/RAF pathway alterations, known as RPAs, are present in up to 80% of analyzed cases. Because driver alterations in RPA pathway genes such as EGFR, BRAF, ALK, RET, ROS1, and KRAS can be targeted by small molecule inhibitors, which significantly prolong survival, clinicians now routinely use such inhibitor drugs as part of their precision medicine treatments for LUAD patients.
The team’s WGS analysis of tumors with no RPA alterations implicated deletions targeting STK11 or KEAP1, genetic alterations targeting promoters, and structural genomic instability as possible factors in the pathogenesis of this important subset of cancers, also known as “oncogene-negative” LUADs.
“Lung adenocarcinoma is unique among cancer types in the fact the majority of cases can be mapped to an oncogenic driver. We were interested to see if WGS could help us fill out the rest of the ‘pie chart’ through more comprehensive detection of variants than was possible in previous whole-exome-focused studies,” noted Dr. Imielinski, who holds a joint appointment as Assistant Professor of Computational Genomics and Assistant Professor of Pathology and Laboratory Medicine at Weill Cornell Medicine. “We uncovered several classes of mutations detectable only with WGS, including structural and noncoding variants, that may drive RPA-negative LUAD. However, we found that this last of slice of the ‘pie chart’ is heterogenous and will require many more samples to characterize. These studies will be useful to expand molecular-guided decision-making to all lung adenocarcinoma patients and make a strong, practical case for WGS in the precision oncology clinic.”
The study team is a working group within the National Cancer Institute’s The Cancer Genome Atlas (TCGA) program, and draws on the tumor dataset collected in that atlas for its analyses. Dr. Imielinski led the study together with co-senior authors Matthew Meyerson, MD, PhD, of Dana Farber Cancer Institute/Harvard Medical School and Ramaswamy Govindan, MD, of Siteman Cancer Center/Washington University School of Medicine. Xiaotong Yao, a PhD student in the Imielinski Lab, is co-first author of the study along with Jian Carrot-Zhang, PhD, of Dana Farber Cancer Institute/Harvard Medical School and Siddhartha Devarakonda,MD, of Washington University School of Medicine. The whole-genome sequencing data processing and analysis for this work was led by Nicolas Robine, PhD, Director, Computational Biology, NYGC, with major contributions from the NYGC Computational Biology team. Additional NYGC co-authors on the study include Aditya Deshpande, PhD student, Imielinski Lab, Michael Zody, PhD, Scientific Director, Computational Biology; Minita Shah, Bioinformatics Scientist; Jennifer Shelton, Bioinformatics Programmer; and Kanika Arora, former Bioinformatics Scientist.
Read more about the research and its findings here.


