NYGC Associate Member Evan Eichler, PhD, NYGC Scientific Director of Computational Biology Michael Zody, PhD, and members of the NYGC Bioinformatics and Software Engineering teams are among the co-authors contributing to this sequencing project that provides unprecedented accuracy against the background of global genetic variation. The new reference will enhance researchers’ ability to make genetic discoveries related to health and disease, especially in groups that have been traditionally under-served by genomics research.
Exactly 20 years after the successful completion of the “Human Genome Project,” an international group of researchers, the Human Genome Structural Variation Consortium (HGSVC), has now sequenced 64 human genomes at high resolution. This reference data includes individuals from around the world, better capturing the genetic diversity of the human species. Among other applications, the work enables population-specific studies on genetic predispositions to human diseases as well as the discovery of more complex forms of genetic variation, as the 65 authors report in the current issue of the scientific journal Science.
In 2001, the International Human Genome Sequencing Consortium announced the first draft of the human genome reference sequence. The Human Genome Project, as it was called, had taken more than eleven years of work and involved more than 1,000 scientists from 40 countries. This reference, however, did not represent a single individual but instead is a composite of humans that could not accurately capture the complexity of human genetic variation.
Building on this, scientists have carried out many sequencing projects over the last 20 years to identify and catalog genetic differences between an individual and the reference genome. Those differences usually focused on small single base changes and missed larger genetic alterations. Current technologies now are beginning to detect and characterize larger differences – called structural variants – such as insertions of several hundred letters. Structural variants are more likely than smaller genetic differences to interfere with gene function.
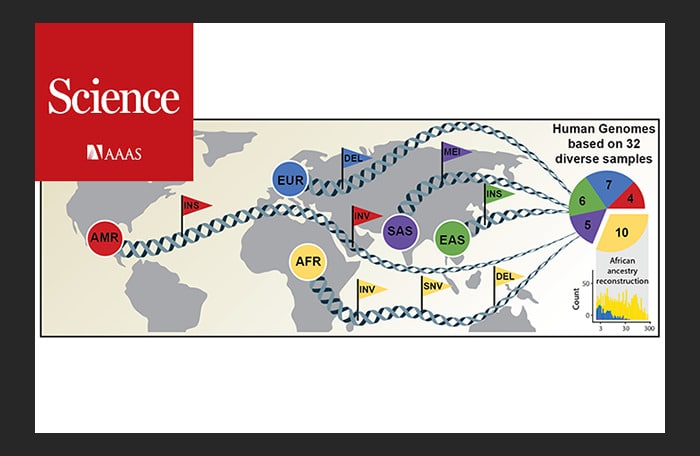
An international research team has now published an article in Science announcing a new, considerably more comprehensive reference dataset obtained using a combination of advanced sequencing and mapping technologies. The new reference dataset reflects 64 assembled human genomes, representing 25 different human populations from across the globe. Importantly, each of the genomes was assembled without guidance from the first human genome and as a result better captures genetic differences from different human populations. The study was led by scientists from the European Molecular Biology Laboratory Heidelberg (EMBL), the Heinrich Heine University Düsseldorf (HHU), The Jackson Laboratory for Genomic Medicine in Farmington, Conn. (JAX), and the University of Washington in Seattle (UW).
“With these new reference data, genetic differences can be studied with unprecedented accuracy against the background of global genetic variation, which facilitates the biomedical evaluation of genetic variants carried by an individual,” emphasizes the co-first author of the study, Dr. Peter Ebert from the Institute of Medical Biometry and Bioinformatics at HHU. “The distribution of genetic variants can differ substantially between population groups as a result of spontaneous and continuously occurring changes in the genetic material. If such a mutation is passed on over many generations, it can become a genetic variant specific to that population.”
NYGC Associate Member Evan Eichler, PhD, Howard Hughes Medical Institute Investigator and Professor in the Department of Genome Sciences, University of Washington School of Medicine, is a co-senior author of the study. NYGC Scientific Director of Computational Biology Michael Zody, PhD, is also a co-author as are NYGC Bioinformatics Scientists Wayne Clarke, PhD, and Anna Basile, PhD; NYGC Senior Bioinformatics Scientist Marta Byrska-Bishop, PhD; NYGC Lead Bioinformatics Scientist André Corvelo, PhD, and NYGC Senior Software Engineer Uday Evani.
“The NYGC team’s whole-genome sequencing of the 1000 Genomes cohort contains all but 1 of the 32 diverse samples used in this study; this data and other NYGC analyses were used to compare the effectiveness of the long-read structural variation detection to short-read detection,” notes Dr. Zody, also a member of the original Human Genome Project. “The paper identifies over 100,000 structural variants, many of them missed entirely by short-read sequencing, and characterizes mechanisms of formation and evolutionary history, as well as highlighting potential impacts on function.”
“Each of these individual genomes is being resolved more completely for a fraction of the price of the first human genome,” says Dr. Eichler, who was also a member of the original Human Genome Project. “We are discovering remarkable differences in genomic organization which have been missed until now, understanding these differences will enhance our ability to make genetic discoveries related to health and disease especially in groups that have been traditionally under-served by genomics research.”
The new reference data provide an important basis for including the full spectrum of genetic variants in so-called genome-wide association studies. The aim is to estimate the individual risk of developing certain diseases such as cancer and to understand the underlying molecular mechanisms. This, in turn, can be used as a basis for more targeted therapies and preventative medicine.
This work might enable further applications in precision medicine. Drug efficacy, for example, can vary between individuals based on their genomes. The new reference data now represent the full range of different genetic variant types and incorporates human genomes of great diversity. Therefore, this new resource might contribute to developing novel approaches in personalized medicine, where the selection of therapies is tailored to a patient’s individual genetic background.
This study builds on a new method published by these researchers last year in Nature Biotechnology to accurately reconstruct the two components of a person’s genome – one inherited from a person’s father, one from a person’s mother. When assembling a person’s genome, this method eliminates the potential biases that could result from comparisons with an imperfect reference genome.


